Alexandra Nelepcu
Farmacist Diriginte


Abordarea temei sindromului mortii subite necesita sensibilitate si tact, data fiind natura sa profund delicata si adesea considerata tabu. Fenomenul, surprinzator si neasteptat, se manifesta diferit intre bebelusi si adulti, reflectand varietatea factorilor de risc si particularitatile biologice specifice fiecarei etape de varsta. In randul celor mai mici, sindromul mortii subite infantile reprezinta o realitate ingrijoratoare, cu cercetari axate pe identificarea si minimizarea factorilor de risc - de la aspecte legate de somn pana la mediul inconjurator.

Sindromul Netherton este o afectiune genetica rara, manifestata printr-o serie de simptome dermatologice complexe si provocatoare. Este o boala ereditara cu transmitere autosomal recesiva, ceea ce inseamna ca pentru a fi afectat, individul trebuie sa mosteneasca o copie defectuoasa a genei implicate de la ambii parinti. Una dintre cele mai distinctive caracteristici ale sindromului este tricorexis invaginata, cunoscuta si sub denumirea de "par de bambus". Acest termen descrie o anomalie a firului de par unde cuticula de la capatul distal al firului se invagineaza in cea proximala, creand o deformare vizibila asemanatoare cu o bucla sau o soseta. Aceasta se datoreaza unei keratinizari defectuoase care apare intermitent de-a lungul firului de par.
Sindromul Fanconi reprezinta o anomalie a functiei tuburilor renale proximali, care conduce la eliminarea excesiva prin urina a elementelor nutritive si minerale esentiale, inclusiv aminoacizi, glucoza, fosfati, acid uric si bicarbonat. Aceasta pierdere excesiva nu doar afecteaza echilibrul hidro-electrolitic si mineral al organismului, dar se manifesta si prin semne clinice precum intarzierea in crestere, poliurie (cresterea volumului de urina), polidipsie (senzatia intensa de sete), deshidratarea si, in cazul copiilor, rahitism – o afectiune ce softenizeaza oasele. La adulti, pierderea progresiva a densitatii osoase poate duce la osteoporoza si osteomalacie, conditii care fragilizeaza oasele si le fac mai susceptibile la fracturi.

Sindromul Gerstmann reprezinta o afectiune neurologica rara, manifestata in urma unor leziuni cerebrale. Aceasta conditie poate rezulta fie dintr-un traumatism cranian, fie din anomalii de dezvoltare. In cazul copiilor, aceasta este desemnata ca sindromul Gerstmann de dezvoltare, iar in termeni medicali este cunoscuta si ca tetrada Gerstmann sau simplu GS. Aceasta patologie subliniaza importanta interconexiunilor dintre diversele zone ale creierului si impactul lor asupra functionarii cognitive si motorii.

Sindromul Klinefelter reprezinta o conditie genetica specifica barbatilor, caracterizata prin prezenta unui numar suplimentar de cromozomi X. In mod obisnuit, barbatii au o configuratie cromozomiala XY, dar cei afectati de acest sindrom prezinta un profil XXY, avand astfel un cromozom X in plus. Aceasta anomalie poate varia, deoarece exista cazuri in care barbatii pot avea doi sau chiar trei cromozomi X suplimentari, si, desi mai rar, un numar mai mare de cromozomi Y.
Sindromul Marcus Gunn, cunoscut si sub denumirea de sincinezia trigemino-oculomotorie, reprezinta una dintre cele mai intalnite forme de sincinezii oculofaciale mostenite. Acest sindrom se manifesta printr-o cadere a pleoapei superioare, variind ca intensitate, si prin miscari involuntare ale aceleiasi pleoape in timpul efectuarii unor actiuni cum ar fi mestecarea sau suptul. Aceasta conditie este de obicei observabila imediat de la nastere, cand parintii pot observa miscarile neobisnuite ale pleoapei copilului lor in timpul hranirii.
Sindromul Horner este o afectiune neurologica deosebit de rara, manifestandu-se prin simptome specifice la nivelul ochiului si ale zonei adiacente de pe o singura parte a fetei. Aceasta conditie semnaleaza prezenta unei probleme nervoase mai profunde, avand multiple cauze potentiale – de la probleme ale arterei carotide pana la prezenta unor tumori la nivelul plamanilor. Identificarea timpurie si tratamentul adecvat sunt esentiale pentru gestionarea eficienta a sindromului. Simptomele caracteristice includ lasarea pleoapei superioare, micsorarea diametrului pupilei, roseata ochiului si absenta transpiratiei pe partea afectata a fetei.
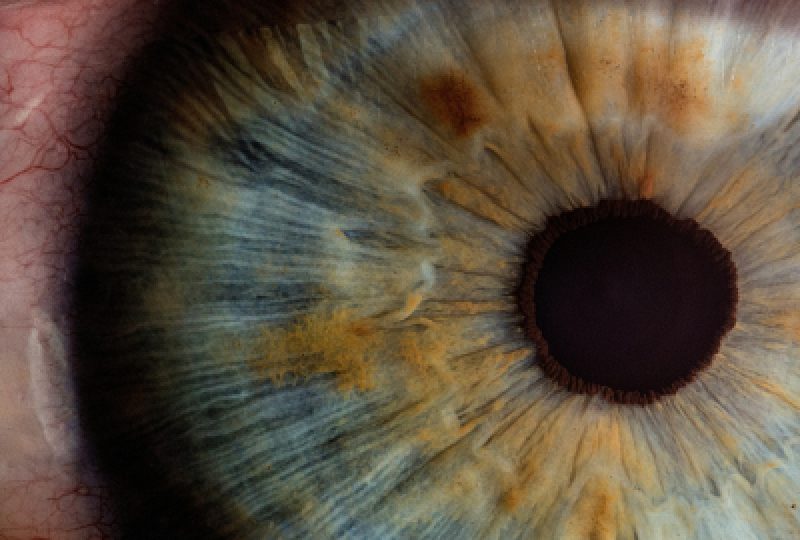
Sindromul Adie, denumit si sindromul Holmes-Adie, reprezinta o anomalie neurologica caracterizata prin afectarea controlului autonom al pupilei, ca urmare a degenerarii ganglionului ciliar. Aceasta afectiune a fost identificata si descrisa pentru prima data in 1931 de catre neurologii William John Adie si Sir Gordon Morgan Holmes, dupa care a fost numita. Principalele semne ale sindromului includ dilatarea anormala a uneia sau ambelor pupile, paralizia partiala a muschilor oculari si o reactie lenta sau absenta a pupilei la lumina. In situatiile in care se observa doar simptomele legate de pupile, afectiunea poarta numele specific de "pupila tonica a lui Adie".
Neuropatia sau afectarea nervilor poate rezulta dintr-o gama larga de afectiuni, cum ar fi diabetul si chiar tratamente precum chimioterapia. De fapt, neuropatia, care este uneori denumita neuropatie periferica, nu este o singura stare de sanatate, ci mai degraba un termen folosit pentru a descrie o serie de probleme de sanatate care implica deteriorarea nervilor periferici, precum si simptomele acestor probleme. In timp ce grupul de afectiuni este ireversibil, puteti lua masuri pentru a ajuta la prevenirea neuropatiei sau a gestiona-o prin dieta, stil de viata si tratament.
